MedTech Europe supports members in navigating the regulatory system. Our training and education resources include videos, infographics, position papers, workshops and direct engagement with members and national industry associations.
The system for ensuring that medical devices meet the necessary standards, specifications, quality, safety and performance requirements is complex. Understanding EU legislation, they key regulatory actors and the essential steps in the CE marking system is vital to bringing products to market.

MedTech Europe supports members in navigating the regulatory system. Our training and education resources include videos, infographics, position papers, workshops and direct engagement with members and national industry associations.
This flowchart has been prepared by MedTech Europe as a ‘high-level overview’ of the requirements of the Medical Devices Regulation. It is intended for informational purpose only and should not be construed as legal advice for any particular facts or circumstances. MedTech Europe reserves the right to change or amend the flowchart or any parts thereof at any time without notice.
This flowchart has been prepared by MedTech Europe as a ‘high-level overview’ of the requirements of the Medical Devices Regulation. It is intended for informational purpose only and should not be construed as legal advice for any particular facts or circumstances. MedTech Europe reserves the right to change or amend the flowchart or any parts thereof at any time without notice.
DownloadThis flowchart has been prepared by MedTech Europe as a ‘high-level overview’ of the requirements of the Medical Devices Regulation. It is intended for informational purpose only and should not be construed as legal advice for any particular facts or circumstances. MedTech Europe reserves the right to change or amend the flowchart or any parts thereof at any time without notice.
DownloadIn the European Union, medical technologies are tightly regulated by laws that govern the safety and performance of devices across their lifetime, pre- and post-market. In the coming years, the European medical technology sector will transition from being regulated under the current medical devices directives to two new regulations. This will bring changes to how products are classified.
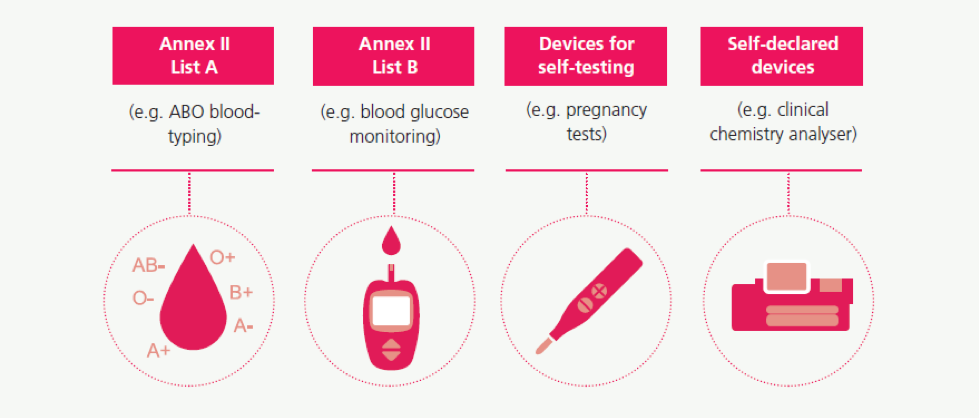
Classification of IVDs determines the level of involvement by a third party (the “Notified Body”) in assessing IVDs both pre- and post-market. This level of control is generally relative to the risk of an erroneous result from the assay. Under the IVD Directive, IVDs are classified into four classes following a positive list approach.
Under the IVD Regulation, all IVDs will be classified under a new risk-based classification system according to the risk the device poses to the health of the public and or an individual as result of an incorrect test result. All IVDs will be classified under class A, B, C or D, with class D being the highest risk class.
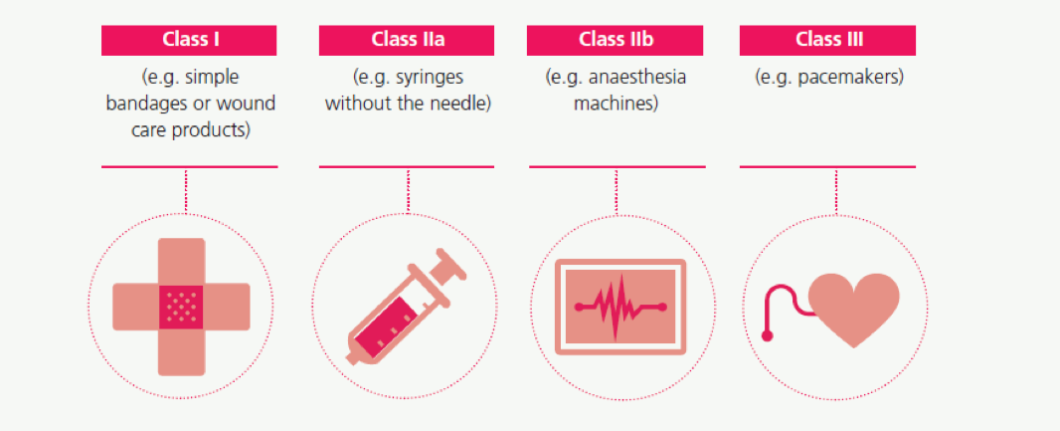
Classification of medical devices drives many pre- and post-market requirements. Due to the large variety of products, the level of control made by a third party (the “Notified Body”) before placing them in the market depends on the level of impact on the human body that their use might imply. The same Notified Body is involved post-market approval to ensure the continued safety and performance of medical devices. Under the MD Directive, MDs are classified into four classes following a risk-based classification system.
Under the new MD Regulation, the risk-based classification system contained in the current Directives has been maintained, although some changes/additions have been introduced. The principle is the same: to link the class of the device to the potential risk posed to the health of the public and an individual resulting from a fault in the functioning of the product. All MDs are classified under class I, IIA, IIB or III, with class III being the highest risk class.
Manufacturers of medical technologies must comply with the requirements of all applicable EU legislation and affix a CE mark to their product before placing it on the market. Notified Bodies are independent certification organisations that determine whether a product meets the relevant requirements for CE marking.
For more training materials and videos, you can visit our YouTube channel. Head over to our dedicated playlist by clicking on the link below.
The MDR/IVDR revision marks a pivotal moment for the EU medtech sector. Read MedTech Europe's full position on the revision.



