Our members are currently managing four- and five-year transition periods to the new regulations. This presents considerable uncertainties that need to be reduced through, for example, practical explanations of what companies can and cannot do over the coming years. It is also essential that we flag our concerns about the feasibility of the transition periods to the authorities.

Transition time: In vitro diagnostics
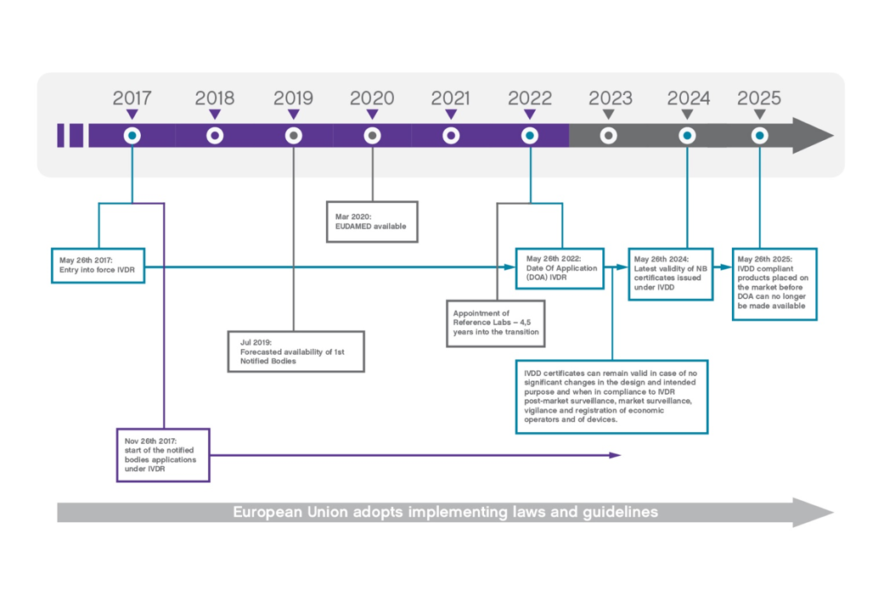
From May 2017 to May 2022, IVDs will transition from being CE marked under the current IVD Directive 98/79/EC to being CE marked under the new IVD Regulation (EU) 2017/746.
However, the transition may last until at least May 2024 for those IVDs that are certified by a Notified Body under the current Directive. No new certifications under the current Directive can take place after 26 May 2022.
EU Member State authorities and Notified Bodies will continue to oversee the EU market, ensuring that all IVDs are fit for purpose and reliably provide information to be used for diagnostic purposes, regardless of whether they are CE marked under the current Directive or the Regulation.
An IVD is not inferior simply because it is CE marked under the current Directive instead of under the new Regulation.
Regulatory documentation – such as Declarations of Conformity, certificates, labels and instructions for use – issued under the current Directive, may remain valid until approximately May 2024.
This valid documentation may therefore remain in circulation, in parallel with new regulatory documentation issued under the new Regulation. Regulatory documents, issued under the new Regulation, may have additional content and different formatting, compared to similar documents issued under the current Directive. This is, again, acceptable and in line with EU legislation.
-
Are IVDs CE marked under the Directive still fit for market circulation?
Are IVDs CE marked under the Directive still fit for market circulation?
Yes, all validly CE marked IVDs can continue to be placed on the EU market during the transition and until the related certificate (for IVDs under Notified Body supervision) expires. It is important to note that there will be a period of up to approximately seven years, during which time certain IVDs CE marked under the Directive, and those CE marked under the new Regulation, will simultaneously (and lawfully) be in circulation.
Simply because an IVD is CE marked under the current Directive does not mean it is exempt from the requirement to be fit for purpose and reliably provide information to be used for diagnostic purposes. In addition, IVDs CE marked under the current Directive will still be subject to an extensive vigilance process to ensure that any problems which may arise are rapidly identified and corrected.
Transition time: Medical Devices
From May 2017 to May 2021, Medical Devices will transition from being CE marked under the two current (and separate) Medical Devices Directives – Directive 93/42/EEC and Directive 90/385/EEC – to being CE marked under the new Medical Devices Regulation (EU) 2017/745.
The system for ensuring that medical devices meet the necessary standards, specifications, quality, safety and performance requirements is complex. This is reflected in the several years given to allow a smooth transition from the Directives to the Regulation.
The transition may last until at least May 2024 for those devices that are certified by a Notified Body under the current Directives. No new certifications under the current Directives can take place after 26 May 2021.
EU Member State authorities and Notified Bodies will continue to oversee the EU market, ensuring that all medical devices are safe and perform as intended, regardless of whether they are CE marked under the current Directives or the new Regulation.
A medical device is not inferior simply because it is CE marked under one of the current Directives instead of under the new Regulation.
Regulatory documentation – such as Declarations of Conformity, certificates, labels and instructions for use – issued under the current Directives, may remain valid until up to approximately May 2024.
-
Are medical devices CE marked under the Directives still fit for market circulation?
Are medical devices CE marked under the Directives still fit for market circulation?
Yes, all validly CE marked medical devices can continue to be placed on the EU market during the transition and until the related certificate (for products under Notified Body supervision) expires. It is important to note that there will be a period of up to approximately seven years, during which time certain devices CE marked under the Directives, and those CE marked under the new Regulation, will simultaneously (and lawfully) be in circulation.
The road ahead
A requirement for all key stakeholders in the process is education about, and adaptation to, the new Regulation. For example:
• EU Member State authorities will need to effectively monitor the new vigilance system, which relies on new tools yet to be made available, such as the new Eudamed database.
• Notified Bodies will need to be assessed and designated under the new Regulation before they can conduct audits of manufacturers and of medical devices.
• Manufacturers must classify their products appropriately and ensure that all product documentation and evidence of compliance is made available and conforms with the new Regulation.
These changes will require considerable time and effort. Everyone involved has a lot of work to do before they can comply with the new Regulations.
For more information please contact MedTech Europe: [email protected]
-
Why does it take so long to transition from the current Directives to the new Regulation? If it is the law everyone must follow it, right?
Why does it take so long to transition from the current Directives to the new Regulation? If it is the law everyone must follow it, right?
Yes, everyone must transition to the new Regulation. But to do so, it must be possible to follow the law. The Regulation adds some new requirements that are intended to be phased in over time. Detailed secondary legislation, containing important implementation details, will be prepared and published by the European Commission over several years. This will address precisely how key provisions of the new Regulation will work. For example, further detailed information will be available regarding how the new European Database on Medical Devices (Eudamed) functions, and which expert panels may support the certification of certain high-risk medical devices.



